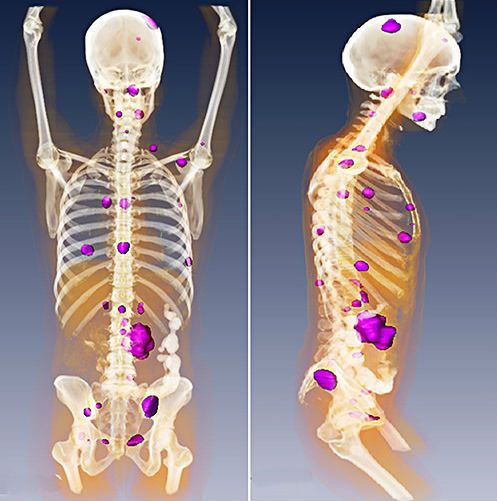

Cancerous tumors must evade immune destruction to survive. They can achieve this by preventing the immune cells from gaining access to the tumor (“immune cold”) or by tipping the balance of immunoactivating and immunosuppressing signals toward immunosuppression. Immunosuppression means that, even if the immune cells penetrate the tumor, they cannot kill the cancer. The cytotoxicity of the immune response is suppressed.
A new study published in Science Translational Medicine shows that injecting two immunostimulating agents into one tumor not only triggered a cytotoxic immune response toward the injected tumor but also a similar response toward metastasized or distant tumors. Particularly interesting is the immunostimulating agents, both of which in a non-cancer context would be considered as stimulating or enhancing the innate immune response to infection. The authors call the treatment “in situ vaccination.” However, contrary to a true vaccine, the treatment does not include any tumor molecules that serve as tumor antigens to teach the immune cells how to recognize the cancer cells. Instead, the treatment involves injecting two agents that could be considered more similar to vaccine adjuvants, the reagents that boost the immune response not train the immune system to recognize a specific antigen. This strategy is highly advantageous, because the tumor antigens are typically unknown. Thus, this treatment relies on enhancing the intratumor immune response, which then recognizes the antigens in the tumor and mounts a systemic antitumor response specific for that type of tumor.
One of the agents in the injected treatment is a short synthetic DNA sequence called CpG oligodeoxynucleotide (CpG ODN), which resembles the microbial DNA and is recognized by the receptor TLR9. TLR9 is found in several cells of the immune system, including NK cells (a type of cytotoxic innate immune cell), B cells (antibody-producing cells), plasmacytoid dendritic cells (infection-fighting cells), macrophages (producers of pro- or anti-inflammatory signals and removers of dead cells, debris, and infectious agents), and antigen-presenting dendritic cells.
The second immunostimulating agent is an antibody that activates the receptor OX40. OX40 is present on two kinds of T cells, cytotoxic effector T (Teff) cells and immunosuppressive T regulatory (Treg) cells. Activation of OX40 in Teff cells results in the proliferation and enhanced cytotoxicity of these cells. Excessive activation of OX40 in Treg cells kills these cells.
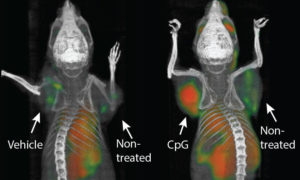
Sagiv-Barfi and colleagues injected CpG ODN into mice with an engrafted tumor and found that the Teff cells in the tumor had an increased amount of OX40 (Figure 1) and that there was a shift in the balance such that there were more Teff cells and fewer Treg cells, compared with the cells in the tumors from the mice that had not been injected. They also noted that human lymphoma patients treated with low dose radiation and intratumoral injection of CpG also had an increase in OX40-positive Teff cells. The authors determined that the CpG ODN-induced increase in OX40-positive Teffs required signals produced by macrophages and dendritic cells. The need for these innate immune cells makes sense, because these are the cells that have TLR9 and respond to CpG ODN. The authors also found the NK cells were required. However, these cells responded to the antibody that activated OX40. However, they did not respond using OX40, the NK cells were activated by the Fc (common part) of the antibody, not the part that recognized OX40.
Using multiple different mouse cancer models, the authors showed that coinjection of CpG ODN and the OX40-stimulating antibody triggered the regression of both the injected tumor and the tumors at distant sites, in many cases curing the mice. For those mice that subsequently developed recurrent tumors, the tumors responded to injection with CpG ODN and OX40-stimulating antibody. The cancer cells did not exhibit resistance. Only CpG ODN had to be injected directly into the tumor to stimulate the antitumor response. The OX40-stimulating antibody could be delivered systemically, but systemic delivery required higher doses than intratumoral injection.
The authors also demonstrated that the antitumor immune response triggered by the combination injection was specific to the type of tumor injected. Mice were with engrafted with two different types of tumors. Injection of one of the tumors only triggered regression of that tumor and any tumors of the same type, but not tumors of the other type. However, if the cells of both types of tumors were engrafted to make a mixed tumor and then that mixed tumor was injected with the immunostimulating agents, the immune system responded to unmixed tumors of each type at distant sites. These data indicated that the antitumor immune response involves multiple tumor antigens, despite the tumor specificity of the response. This is encouraging because human cancers tend to be heterogeneous with cells that have different genetic mutations and potentially different immunogenic profiles.
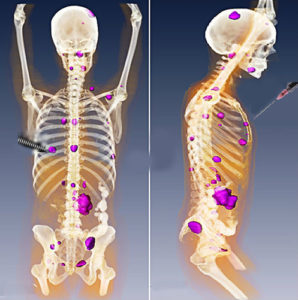
One intriguing aspect of this study is the connection to radiation-treated lymphoma in humans. Radiation is one way to make an immune-cold tumor hot by physically disrupting the tumor and causing death of the tumor cells to expose intracellular proteins and molecules that serve as antigens for the immune system. However, radiation alone has not produced strong antitumor immune responses. Thus, the immune system may require a boost to achieve a durable antitumor response. Indeed, many studies of tumor radiation indicate that similar immune-modulating signals (cytokines) and cellular immune responses are produced as those observed with the intratumor injection of CpG. Like CpG injection, radiation alone is not sufficient to produce a systemic antitumor immune response. This study suggests that, just like combining immune checkpoint inhibitors with radiation may boost the anticancer immune response, so may combining radiation with such immunostimulating agents (Figure 2). More research is necessary to determine which patients and which cancers will best respond to which types of treatment.
Highlighted Articles and Related Reading
Sagiv-Barfi, D. K. Czerwinski, S. Levy, I. S. Alam, A. T. Mayer, S. S. Gambhir, R. Levy, Eradication of spontaneous malignancy by local immunotherapy. Sci. Transl. Med. 10, eaan4488 (2018). Abstract
Van-pouille-Box, S. C. Formenti, S. Demaria, Toward precision radiotherapy for use with immune checkpoint blockers. Clin. Cancer Res. 24, 259-265 (2018). Pubmed
Clinical Trials
CpG ODN
Genes and Proteins
TLR9
Gene https://www.ncbi.nlm.nih.gov/gene/54106
Protein http://www.uniprot.org/uniprot/Q9NR96
OX40
Gene https://www.ncbi.nlm.nih.gov/gene/7293
Protein http://www.uniprot.org/uniprot/P43489
Related Commentary
N. R. Gough, Immunotherapy antagonists and agonists. BioSerendipity (25 May 2017). https://www.bioserendipity.com/2017/05/25/immunotherapy-antagonists-and-agonists/
N. R. Gough, Making immune “cold” tumors hot. BioSerendipity (31 May 2017). https://www.bioserendipity.com/2017/05/31/making-immune-cold-tumors-hot/
N. R. Gough, Understanding immune checkpoint pathways to improve patient response. BioSerendipity (24 May 2017). https://www.bioserendipity.com/2017/05/24/understanding-immune-checkpoint-pathways-to-improve-patient-response/
Cite as: N. R. Gough, Intratumor immunotherapy kills injected and distant tumors. BioSerendipity (1 February 2018). https://www.bioserendipity.com/2018/02/01/intratumor-immunotherapy-kills-injected-and-distant-tumors/