
Defining >56,000 Human Protein Interactions through Mass Spectrometry
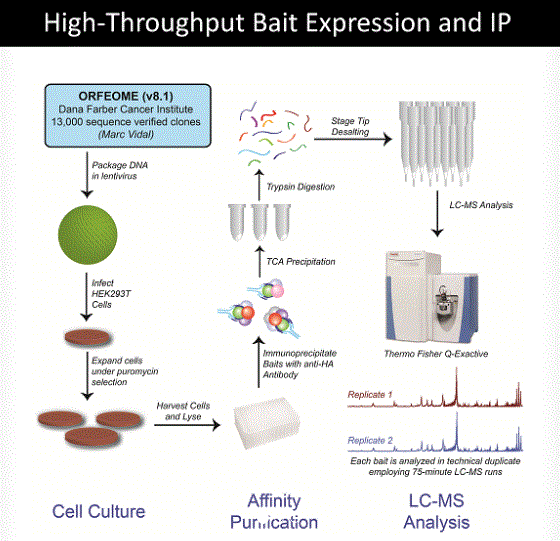
Huttlin and colleagues applied mass spectrometric analysis to affinity purified protein complexes to help build a partial reference map of the human protein interaction space (1). The cDNA encoding the target proteins also engineered to have affinity tags (hemagglutinin and FLAG) were introduced into an easily infected human cell line. The tagged target proteins were expressed and then affinity purified and subjected to mass spectrometric analysis to identify the proteins that associated with the affinity-purified target proteins (Figure 1). This approach identifies both direct and indirect protein interactions that form protein complexes.
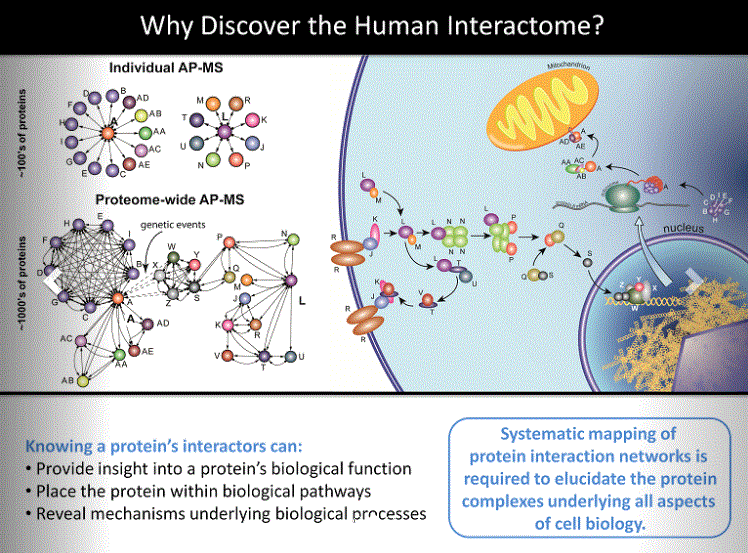
The human protein interaction space still lacks a complete reference set even for just the direct binary interactions (direct interactions between 2 proteins) (2). In the published work of Huttlin and colleagues (1), the pipeline and processes used enable the analysis of 500 proteins encoded in the human genome per month. By combining the analysis of 3,297 proteins in this new pipeline (BioPlex2.0) with a re-analysis of the 2,594 proteins analyzed in the previous version (BioPlex) (3), the results represent one of the largest collections of protein interaction data analyzed through a single methodology: 5,891 proteins targeted for affinity purification to yield 56,553 interactions among 10,961 proteins. After publication of the study, an additional 1,712 proteins were subjected to affinity purification and their binding partners analyzed. All data are included in BioPlex2.0 online (4).
Uncovering Clues to the Uncharacterized Proteome through “Guilt by Association”
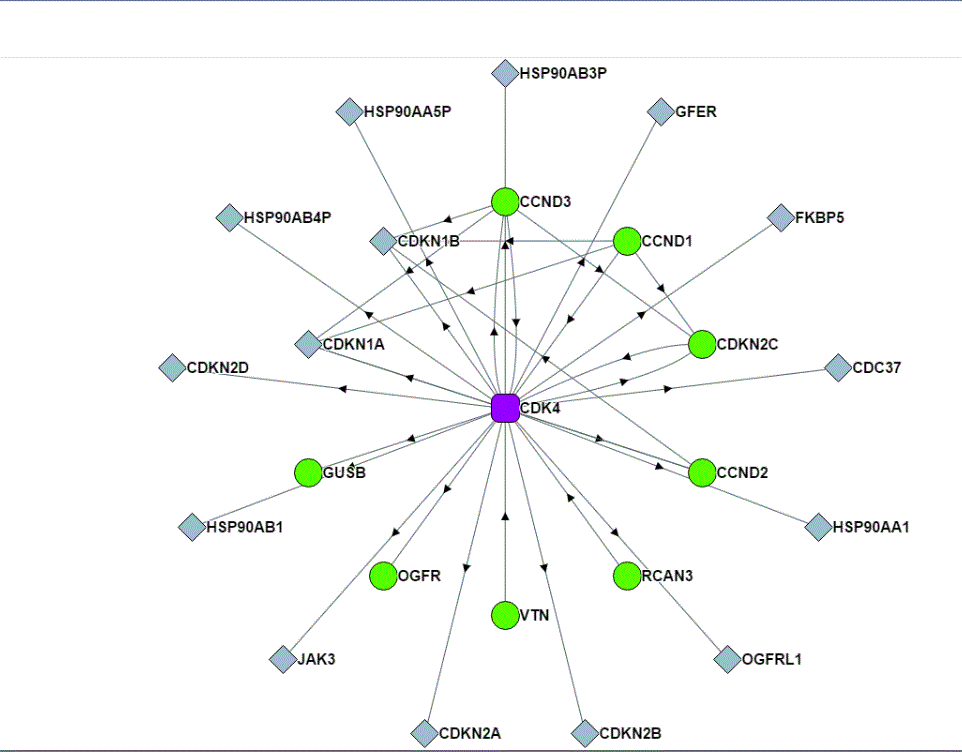
What is the advantage of having such a database, which is still far from complete? There are ~20,000 protein-coding genes in the human genome, and there are allelic variants, alternative splice forms, bioactive cleavage products, and other variants in the human proteome. Even the direct binary interaction space may be ~150,000 interactions when only considering a single protein product from each gene (2). So, the 56,553 interactions, which consist of both direct and indirect interactions and is limited to those interactions that are not disrupted by the conditions used for affinity purification and that can occur in the transfected cells, really represents a very small number of the total number of interactions in all human cells. Yet, with a database of this size, there is statistical power and the power of inference, a kind of proteinacious “guilt by association,” which can provide clues about proteins of unknown function (Figure 2).
One example of such inferential knowledge is the prediction of the subcellular localization of a protein using the localization assigned to the proteins with which the uncharacterized protein affinity purified. Another is the prediction of protein function from the functions assigned to the proteins in the same affinity-purified complex. This manner of assigning protein properties and function relies on accurate ontology-based annotation of the characterized proteins in the purified complex. Huttlin and colleagues (1) used ontology data from UniProt (5) for subcellular localization and Gene Ontology (GO) (6, 7) for function. This type of inferential knowledge is a powerful way to guide experiments and generate hypotheses.
Discovering Proteins Associated with Disease
A biomedically important application of this type of proteomic data is in understanding disease, generating hypotheses about disease mechanisms, and identifying potentially new targets for intervention. Proteins that are functionally related—work in the same regulatory pathway, act in the same biochemical process, contribute to the same subcellular structure—often form interconnected protein clusters. With the data in BioPlex2.0, the authors used computational analysis to assign the proteins into 1,320 clusters, which they call “communities.” These communities had as few as three proteins or as many as 76 proteins and exhibited 286 different network topologies. To identify new disease associations, the authors added the data from DisGeNET (8), which annotates genes with diseases, onto the protein communities. This approach identified 442 protein communities that associated with 2,053 diseases. Not only did this reveal that different diseases displayed different distributions among the communities, which provides insights into the complexity of different diseases and reveals potential strategies or challenges to therapeutic intervention, but combining disease information with the protein network data also enabled the generation of testable hypotheses about how altered protein interactions may contribute to disease. Thus, even a partial protein interaction dataset has the power to produce medically important discoveries.
Highlighted Articles and Websites
- E. L. Huttlin, R. J. Bruckner, J. A. Paulo, J. R. Cannon, L. Ting, K. Baltier, G. Colby, F. Gebreab, M. P. Gygi, H. Parzen, J. Szpyt, S. Tam. G. Zarraga, L. Pontano-Vaites, S. Swarup, A. E. White, D. K. Schweppe, R. Rad, B. K. Erickson, R. A. Obar, K. G. Guruharsha, K. Li, S., Artavanis-Tsakonas, T. P Gygi, J. W. Harper, Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509 (2017). PubMed
- M. Vidal, How much of the human protein interactome remains to be mapped? Sci Signal 9, eg7 (2016). PubMed
- E. L. Huttlin, L. Ting, R. J. Bruckner, F. Gebreab, M. P. Gygi, J. Szpyt, S. Tam, G. Zarraga, G. Colby, K. Baltier, R. Dong, V. Guarani, L. Pontano Vaites, A. Ordureau, R. Rad, B. K. Erickson, M. Wühr, J. Chick, B. Zhai, D. Kolippakkam, J. Mintseris, R. A. Obar, T. Harris, S. Artavanis-Tsakonas, M. E. Sowa, P. De Camilli, J. A. Paulo, J. W. Harper, S. P. Gygi, The BioPlex network: A systematic exploration of the human interactome. Cell 162, 425–440 (2015). PubMed
- BioPlex (accessed 23 June 2017) http://bioplex.hms.harvard.edu
- UniProt (accessed 23 June 2017) http://www.uniprot.org/
- Gene Ontology (accessed 23 June 2017) http://www.geneontology.org/
- AmiGO (accessed 23 June 2017) http://amigo.geneontology.org/amigo
- DiGenNet (accessed 23 June 2017) www.disgenet.org
Cite as: N. R. Gough, The Power of Proteomics in Building the Human Protein Interaction Network. BioSerendipity (23 June 2017) https://www.bioserendipity.com/2017/06/23/the-power-of-proteomics-in-building-the-human-protein-interaction-network/.
Don’t miss a post! Get an email when new content is posted: Sign Up