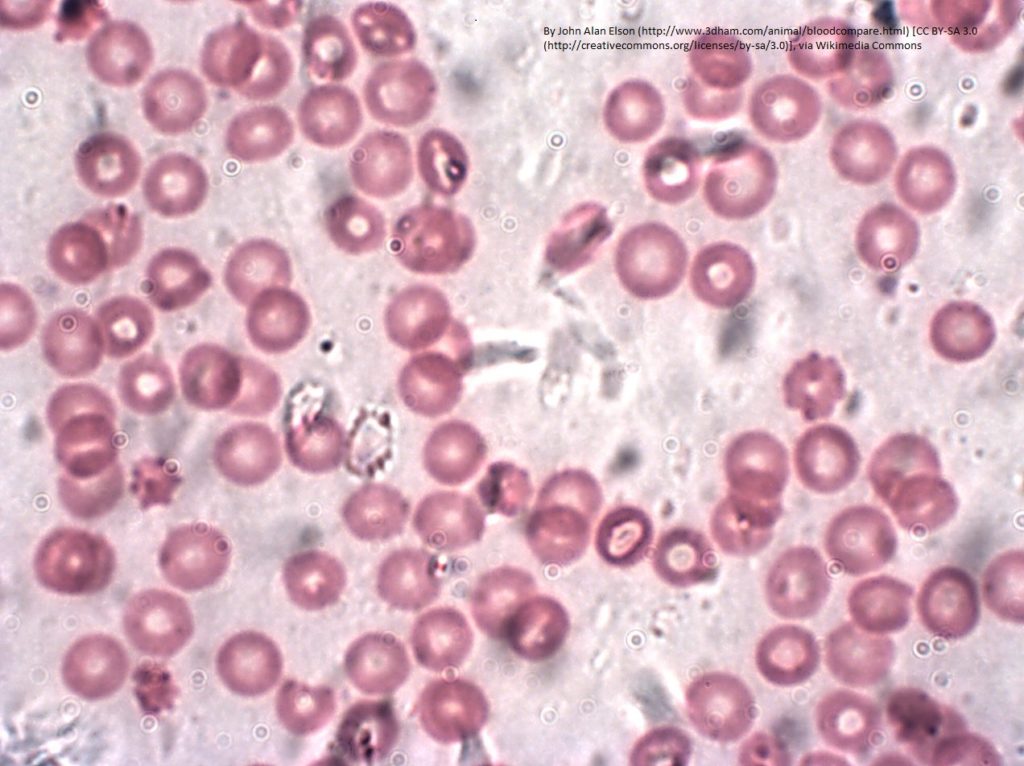

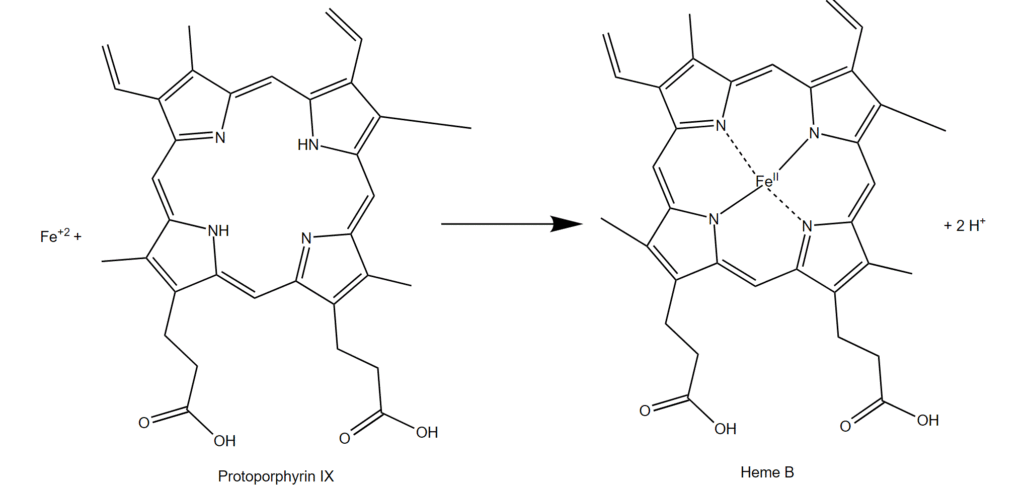
Ferrochelatase in heme biosynthesis
Ferrochelatase (FECH) is an enzyme found in mitochondria that adds iron ions (Fe2+) into protoporphyrin to produce heme (Figure 1). This biochemical reaction is critical for cell health. The process is tightly controlled so that cells produce the amount of heme needed and do not accumulate too much of the heme precursor. The best-known protein that contains heme is hemoglobin, which is the oxygen-carrying protein in red blood cells. The ability of hemoglobin to carry oxygen depends on the iron in the heme cofactor. Heme is also present in proteins that function in mitochondrial energy production, in proteins that function in biochemical synthesis and degradation processes, and in proteins that detoxify hydrogen peroxide in cells.
Mutations in FECH that impair activity result in the disease erythropoietic protoporphyria (EPP). The main symptom of EPP is extreme sensitivity to light (photosensitivity). This photosensitivity occurs because skin cells accumulate protoporphyrin. Protoporphyrin is a type of molecule called a porphyrin. As such, protoporphyrin absorbs light and then releases the energy as heat and forms a reactive oxygen molecule that causes cellular damage. This light-induced process causes the burning sensation and skin irritation that are the main symptoms of EPP. In rare cases, the excessive protoporphyrin causes damage to the liver and gall bladder (hepatobiliary disease).
Perhaps surprisingly given the importance of heme for cells, several FDA-approved drugs interfere with heme production. Some (griseofulvin) inhibit ferrochelatase directly; others (deferoxamine) inhibit ferrochelatase indirectly by chelating iron. Others (5-ALA) overwhelm the ability of ferrochelatase to make heme by producing too much of the substrate protoporphyrin. As highlighted in the commentary of 10 July 2017, preclinical studies indicate that ferrocheletase inhibitors may be useful in treating vascularization-associated loss of vision. The iron-chelating drugs are used in cases of iron poisoning to clear excess iron from the body. The photosensitizing properties, which result in light-induced cellular damage, of 5-ALA treatment are used to treat neovascularization-associated loss of vision and some cancers. The light-absorbing properties of the protoporphyrin that accumulates in response to 5-ALA are under investigation for diagnosis of solid cancers and identifying tumor margins.
A regulatory pathway for heme biosynthesis
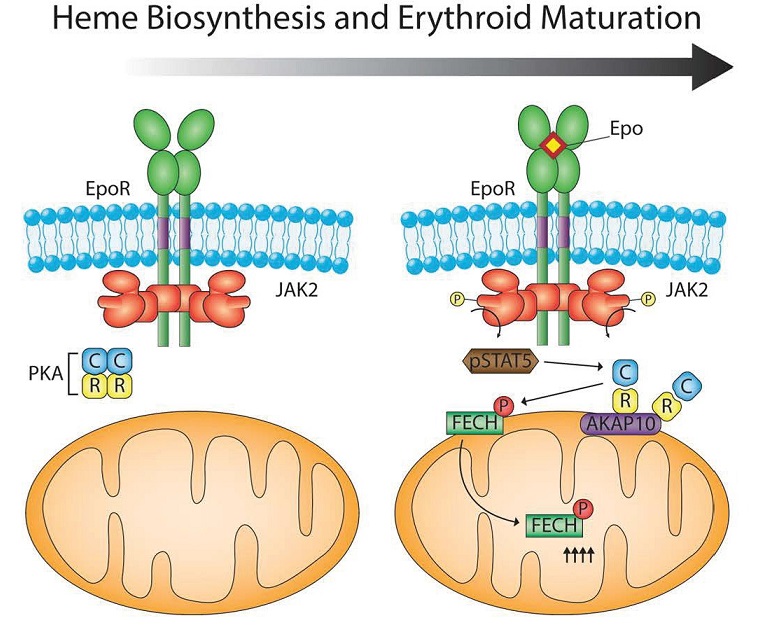
The regulatory and catalytic subunits of the enzyme protein kinase A (PKA) and scaffold protein AKAP10, which functions as a PKA-anchoring protein, were among the mitochondrial proteins that were more abundant in the differentiated MELs. Follow-up analysis showed that PKA localized to the outer membrane of the mitochondria and that this localization depended on AKAP10 in the differentiating MELs. Expression of the gene AKAP10 increases during red blood cell differentiation and the gene is stimulated by the transcription factor GATA1, which is activated by erythropoietin (EPO). EPO and GATA1 are critical for red blood cell development. Thus, EPO stimulates the GATA1-induced expression of AKAP10, which increases AKAP10 abundance. AKAP10 localizes to the mitochondria and recruits PKA to the mitochondria. These events provide the regulatory connection between cytokine that controls red blood cell development, the transcriptional processes that induce red blood cell formation, and the increase in PKA at the mitochondria in the differentiated MELs.
The next questions are
Is this AKAP10-targeted PKA important for hemoglobin production?
If so, what is the relevant target of PKA in this process?
Knocking out the gene for AKAP10 in the MEL cells prevented the recruitment of PKA to the mitochondria and impaired the produce of hemoglobin, and knocking down AKAP10 in zebrafish embryos resulted in anemia due to impaired hemoglobin synthesis. These assays confirm the importance of AKAP10 in hemoglobin production.
Analysis of the amino acid sequences of the mitochondrial enzymes involved in hemoglobin biosynthesis identified FECH as the only one of the four with a sequence that PKA would recognize and phosphorylate. Biochemical assays confirmed that purified PKA phosphorylated purified FECH at the residue in FECH that is the PKA consensus phosphorylation site (Thr116 in humans and Thr115 in mice). The differentiating MEL cells lacking AKAP10 had less phosphorylated FECH, consistent with AKAP10 recruiting PKA to the mitochondria where it phosphorylates FECH.
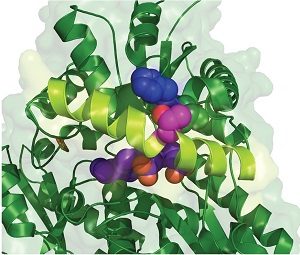
Inspection of the crystal structure of FECH showed that Thr116 was close to the active site and predicted that phosphorylation would alter FECH activity (Figure 3). Indeed, phosphorylated human FECH had increased activity compared with the nonphosphorylated form. Mice with FECH with a mutation at Thr115 that blocked phosphporylation had less hemoglobin in their red blood cells and the cells had excess amounts of the heme precursor protoporphyrin.
PKA is not always active. Indeed, under nonstimulated conditions, PKA is kept in an inactive complex with the regulatory subunits. Stimuli that increase the activity of the enzyme adenylyl cyclase result in the increased production of the molecule cAMP, which binds the regulatory subunits letting the catalytic subunits of PKA become active.
So, the next question is
What signal activates PKA in the differentiating red blood cells?
The first candidate to test is EPO. When EPO binds its receptor EPOR (Figure 4), which is associated with the kinase JAK2, JAK2 is activated. EPO is used clinically to treat anemia, like that associated with chemotherapy. Through JAK2, EPO stimulates the transcription factor STAT5, which increases expression of the gene for the transferrin receptor so that red blood cell precursors will take up more iron from the circulation. Surprisingly, mice that overexpress EPO do not have iron overload and toxicity in the developing red blood cells. Chung and colleagues provide an answer to how iron overload is avoided: EPO also stimulates the phosphorylation of FECH at the PKA, thereby increasing the incorporation of iron into heme and hemoglobin synthesis. This phosphorylation of FECH (and another PKA target) depended on the kinase JAK2. Instead of involving a change in cAMP production, the authors found that JAK2 stimulates the phosphorylation of STAT5, which in addition to moving into the nucleus to stimulate gene expression, also interacted with the catalytic subunit of PKA. Thus, the interaction with STAT5 may induce the release of the PKA catalytic subunits from the regulatory subunits, letting PKA phosphorylate its targets. This STAT5-mediated activation may not require an increase in cAMP.
Remaining questions
How does an enzyme in the inside of the mitochondria get activated by phosphorylation by an enzyme on the outside of the mitochondria?
Does EPO also stimulate or alter the binding properties of the proteins that move mitochondrial proteins to different compartments of the mitochondria?
Does only newly synthesized FECH become phosphorylated only prior to entry into the mitochondria?
What dephosphorylates FECH once sufficient heme has been generated to reset the system to baseline?
FECH is an enzyme that functions in the matrix (middle part) of the mitochondria. How does PKA, which is anchored to the outer membrane phosphorylate a protein in the matrix of the mitochondria. Mitochondria have complex protein movement machines that transport proteins into the various parts of the mitochondria and out of the mitochondria. The authors speculate that before FECH is transported into the matrix, it is phosphorylated by PKA at the outer surface (Figure 5).
This study highlights how incredibly complicated mitochondria are, both in terms of their functions and in terms of their composition. The proteins that are found in or associated with mitochondria are dynamic, interacting with other proteins, moving from one part of the mitochondria to another, and becoming posttranslationally modified so that their activity can be properly regulated.
Highlighted Article
J. Chung, J. G. Wittig, A. Ghamari, M. Maeda, T. A. Dailey, H. Bergonia, M. D. Kafina, E. E. Coughlin, C. E. Minogue, A. S. Hebert, L. Li, J. Kaplan, H. F. Lodish, D. E. Bauer, S. H. Orkin, A. B. Cantor, T. Maeda, J. D. Phillips, J. J. Coon, D. J. Pagliarini, H. A. Dailey, B. H. Paw, Erythropoietin signaling regulates heme biosynthesis. eLife 6, e24767 (2017). PubMed
Related BioSerendipity Commentary
N. R. Gough, Drug Repurposing: Preventing Blindness with an Antifungal. BioSerendipity (10 July 2017). https://www.bioserendipity.com/2017/07/10/drug-repurposing-preventing-blindness-with-an-antifungal/
Cite as: N. R. Gough, The Central Role of Mitochondria in Red Blood Cells. BioSerendipity (14 July 2017). https://www.bioserendipity.com/2017/07/14/the-central-role-of-mitochondria-in-red-blood-cells/